
揭秘AI蛋白质预测“大考”:谁能脱颖而出?
2025-07-25 15:30:51 阅览:388
【导语】随着AI技术的飞速发展,蛋白质结构和功能的预测工具如雨后春笋般涌现。然而,这些工具的有效性高度依赖于庞大的蛋白质数据集。本文将通过斐波那契数列的比喻,揭示AI预测蛋白质结构和功能的本质。同时,针对当前数据库中信息可信度低的问题,介绍上海交(jiāo)通(tōng)大(dà)学(xué)开(kāi)发(fā)的(de)VenusMutHub评(píng)测(cè)平(píng)台(tái),该(gāi)平(píng)台(tái)通(tōng)过(guò)严(yán)格(gé)筛(shāi)选(xuǎn)临(lín)床(chuáng)验(yàn)证(zhèng)数(shù)据(jù),对(duì)AI工(gōng)具(jù)进(jìn)行(xíng)“大(dà)考(kǎo)”。考(kǎo)试(shì)结(jié)果(guǒ)揭(jiē)示(shì)了(le)AI工(gōng)具(jù)的(de)局(jú)限(xiàn)性(xìng),并(bìng)为(wèi)从(cóng)业(yè)者(zhě)提(tí)供(gōng)了(le)选(xuǎn)择(zé)和(hé)使(shǐ)用(yòng)AI工(gōng)具(jù)的(de)明(míng)确(què)思(sī)路。未(wèi)来(lái),AI蛋(dàn)白(bái)质(zhì)预(yù)测(cè)的(de)进阶之路仍需不断探索,期待“全能AI”的出现,为蛋白质预测领域带来新突破。
随着AI模型不断进步,用于预测蛋白质结构和功能的计算机工具如雨后春笋版不断涌现。然而,这些工具通常依赖于一个极(jí)大(dà)的(de)(高(gāo)通(tōng)量(liàng)的(de))蛋(dàn)白(bái)质(zhì)数(shù)据(jù)集。简(jiǎn)单(dān)来(lái)说(shuō),让(ràng)AI预(yù)测(cè)蛋(dàn)白(bái)质(zhì)的(de)结(jié)构(gòu)和(hé)功(gōng)能(néng)就(jiù)像(xiàng)玩(wán)“找(zhǎo)规(guī)律(lǜ)”游(yóu)戏(xì)。下(xià)列(liè)数(shù)字(zì),大(dà)家(jiā)一(yī)定(dìng)不(bù)陌(mò)生(shēng):
1,1,2,3,5,8,X,
X=?
你(nǐ)肯(kěn)定(dìng)认(rèn)出(chū)来(lái)了(le),这(zhè)是(shì)著(zhe)名的(de)斐(fěi)波(bō)那(nà)契(qì)数(shù)列(liè)。通(tōng)过(guò)观(guān)察(chá)能(néng)发(fā)现(xiàn),数(shù)列(liè)中(zhōng)的(de)每(měi)一(yī)项(xiàng)都(dōu)等(děng)于(yú)前(qián)两(liǎng)项(xiàng)之(zhī)和(hé),因(yīn)此(cǐ)X=5+8=13。

图库版权图片,转载使用可能引发版权纠纷
要想让AI预测出精准、符合事实的“X”,就得给它输入足量且正确的前置信息,就是数列中X之前的项。基于对这些前置项的学习,AI才能找到规律,给出有意义的预测结果。
但真实的情况是,数据库中经过生化性质检验的结果占比较低,来自临床样本的数据更是少之又少。库中大多数的蛋白质功能标签都来自先前的结构预测工具——也就是说,这些“功能”本身就是现有AI的前辈们推测出来的。这就好比将具有多个规则的数列都混合在一起,还随机插入一些出题人“灵光一现”的数字,再让AI找规律。可想而知,这样找出的“规律”必然与真实的自然规律相去甚远,也远不具备产业转化价值。
破局之策:AI大模型赋能蛋白质功能预测
面对上述难题,上海交通大学的学者们开发出了蛋白质预测AI评测平台VenusMutHub,它就像一场针对AI的“大考”,专门为这些预测工具“打分”,为研究者们提供更精准的AI工具使用思路。
既然有“考试”,那就要先命制“考卷”。由于数据库中的大多数蛋白质数据可信度较低,为了筛选出对产业进步有实际帮助的工具,这张“考卷”只能包含经过临床验证或生化实验确定功能的蛋白质结构数据。开发人员从多个数据库中层层筛选,最终构建了包括527种不同蛋白共计905个蛋白质突变数据集。这个集合包括了蛋白质突变后稳定性、活性、与其他分子结合的亲和力等多方面数据,且均经过实验验证。将这些(xiē)数(shù)据(jù)集投(tóu)喂(wèi)给(gěi)AI工(gōng)具(jù)后(hòu),开(kāi)发(fā)人(rén)员(yuán)对(duì)AI工(gōng)具(jù)给(gěi)出(chū)的(de)数(shù)据(jù)进(jìn)行(xíng)整(zhěng)理(lǐ),然(rán)后(hòu)分(fēn)科(kē)目(mù)对(duì)它(tā)们(men)的(de)表(biǎo)现(xiàn)进(jìn)行(xíng)“打(dǎ)分(fēn)”。
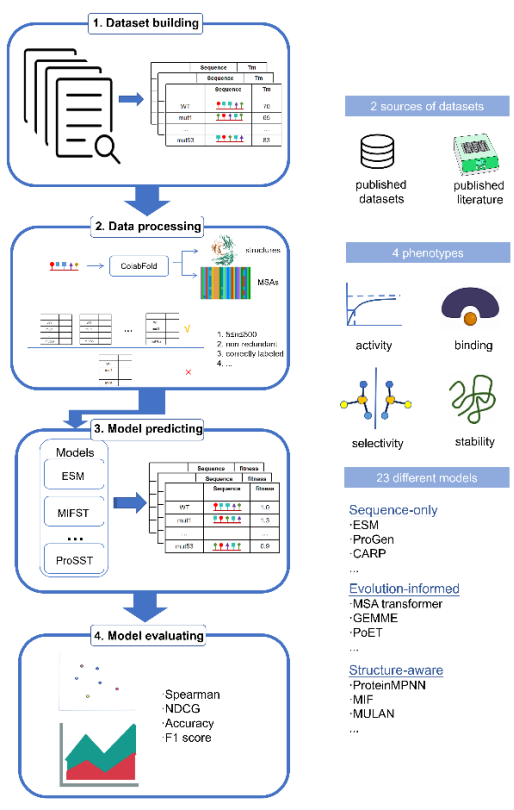
图(tú)片(piàn)来(lái)源(yuán):上(shàng)海(hǎi)交(jiāo)通(tōng)大(dà)学(xué)教(jiào)育(yù)部(bù)科(kē)学(xué)工(gōng)程(chéng)计(jì)算(suàn)重(zhòng)点(diǎn)实(shí)验(yàn)室(shì)官(guān)网(wǎng)
考(kǎo)试(shì)结(jié)果(guǒ)大(dà)揭(jiē)秘(mì):AI工(gōng)具(jù)的(de)“众(zhòng)生(shēng)相(xiāng)”
这(zhè)场(chǎng)考(kǎo)试(shì)的(de)结(jié)果(guǒ)也(yě)相(xiāng)当(dāng)有(yǒu)趣(qù)。开(kāi)发(fā)人(rén)员将“赶考”的AI工具大致分为三个组别:结构预测型(主要关注蛋白质三维结构的预测)、进化信息型(主要关注同一蛋白在不同物种间的序列差异)和纯序列型(主要从氨基酸序列出发进行对比和预测)。
在样本量对预测结果的影响方面,当突变数量高于28个,结构预测型工具的可信度全面领先于其他模型,表现出相当高的可信度。然而,当突变数量小于8个时,所有的模型都无法给出有效结果——全在“胡说八道”。这就像是当数列中给出的已知项不到8个时,所有AI都无法预测出下一项“X”的真面目,但大多数AI会凭借算法“捏造”一个看似合理的答案。
这为AI工具的使用敲响了警钟:目前所有的蛋白质突变预测工具均无法在可靠样本值太小的前提下得出可靠结论,所谓“AI完全取代实验室”“仅凭计算机技术开发药物”是完全错误的,无论多厉害的算法工具都离不开实验室提供的数据支撑,AI技术预测到的结果在大规模投入临床和生产前也必须经过严格的细胞或动物实验验证。
现有多数通用工具对协同效应捕捉有限,已出现少量专门模型尝试解决,但整体准确率仍不理想。开发人员发现,参与检测的AI工具在预测单一位点突变时还比较可靠,但涉及到同时突变两个位点时就再次集体“哑火”了。它们只能识别出简单的叠加作用,即1+1=2。但在自然界中,很多突变之间存在相互协作,会出现1+1>2(正协同)或1+1<2(负协同)的情况,这被AI工具们集体忽略了。
除此以外,AI工具们也像赶考的学生们一样表现出了各种“偏科”现象。有的AI某一科目打分很高,却在另一科目几乎不及格;有的AI平均分看似很高,但却“深一脚浅一脚”,遇到某些蛋白预测精准,另一些却胡说八道;还有的AI看似平均分不太高,但输出均衡,是个成绩稳定的中等生。但不管是哪种AI工具,都不能做到“全才”,总有一个科目得分比较低。

图库版权图片,转载使用可能引发版权纠纷
AI蛋白质预测的进阶之路在何方?
总而言之,这次AI工具的集体“大考”撕下了“AI无所不能”的神话面具,为从业者们提供了明确的思路。对于产业从业者来说,根据研究目的选择适宜的AI工具非常重要,并且至少需要提供8个可靠的突变数据。而对于开发者,如何提升工具在小样本量条件下的预测精确度、教会AI预测多个突变位点对蛋白质结构和功能的影响更为重要。期待有一天,科科满分的“全能AI”能够横空出世,为蛋白质预测领域带来新的突破。
参考文献:
https://doi.org/10.1016/j.apsb.2025.03.028
《生物信息学(第四版)》 陈铭主编 科学出版社
作者:何一文 清华大学本硕,中学教师
审核:李旭 中国科协研究员,中国科学技术大学副教授
出品:
本文封面图片来自版权图库,转载使用可能引发版权纠纷